Nature favours Nonsense
by Mohamed El Brolosy
Throughout an organism’s life-span, cells accumulate mutations as a result of exposure to DNA damage-inducing agents such as UV radiation or mutagenic chemicals. From an evolutionary perspective, survival depends on how fast organisms can adapt to change. If we think of changes in the genome, the fittest organisms will be those that can compensate for harmful genetic mutations, allowing their vital functions to remain intact. A mutated gene that is not compensated for can lead to life-threatening diseases, such as cystic fibrosis, where patients suffer from devastating build-up of mucus in their lungs.
To understand a specific gene’s function, scientists make use of gene targeting technologies to generate mutations in model organisms and investigate the effect of gene loss on the organism. Interestingly, it was observed that many genetically engineered mutant organisms show no obvious observable phenotypic defects. Such observations have resulted in an increased interest in understanding how organisms can adapt to or compensate for mutations, a phenomenon termed ‘genetic robustness’.
One of the proposed mechanisms behind genetic robustness is called ‘genetic compensation’. This involves increased transcription, i.e., production, of other related genes that can functionally compensate for the mutated gene. Genes code for the production of functional proteins in the cell through an intermediate molecule known as RNA and mutations often lead to production of defective RNAs. These are then removed from the cell by the RNA quality control machinery, through a process called degradation, which involves cleaving the mutant RNA into smaller fragments. The most common of such mutations is called nonsense mutations; these cause the protein-production machinery, the ribosome, to terminate protein production prematurely. Studying genetic compensation in zebrafish and mouse at the Max Planck Institute for Heart and Lung research, we found that only mutations that lead to defective RNAs that are subjected to degradation, such as nonsense mutations, could be compensated for, but other mutations, such as those that do not produce a defective RNA, could not (El-Brolosy et al., Nature, 2019).
As said before, mutations can lead to defective RNA, which is then removed from the cell through degradation. We found that the more defective RNA is removed (i.e., the more degradation of mutant RNA), the more the compensating genes become productive. To investigate how the degradation of mutant RNA might trigger genetic compensation, we used the CRISPR/Cas9 system (see discussion about that technology in this issue of Offspring). We induced mutations in several genes in zebrafish that caused the animal to be unable to produce the mutated gene's defective RNA. Because they cannot produce the defective RNA, there is no degradation of defective mutant RNA and thereby we should not expect to observe a compensatory response. Indeed, we found that such mutant animals did not induce a compensatory response to the mutation. More interestingly, these animals displayed developmental defects that were not observed in other animals with mutations leading to production of defective RNAs. Such defective RNAs in these other animals would be subjected to degradation, and trigger a compensatory response, resulting in the lack of developmental defects. For instance, nonsense mutations (which cause RNA degradation) in the egfl7 gene (which controls proper blood vessel development in zebrafish embryos), do not lead to defects in the vasculature’s development (Figure 1), because genetic compensation mechanisms lead to increased production of other related genes that compensate for the defect. However, a zebrafish mutant for the egfl7 gene that do not produce defective RNAs, did not lead to a compensatory response and thereby displayed strong defects in blood vessels development (Figure 1).
Our study has provided evidence for genetic compensation as a gene regulatory mechanism and further increased our understanding of genetic robustness as a whole. Genetic compensation might explain reports of ‘resilient’ healthy individuals harboring mutations without presenting any phenotypic defects or symptoms (Sulem et al., Nat. genetics, 2015; Lek et al., Nature, 2016). Specifically, some studies report that individuals with genetic mutations that cause mutant RNA degradation (i.e., the removal of defective RNA) to cause milder forms of some genetic diseases, such as Marfan syndrome and Beta-Thalassemia intermedia compared to individuals with mutations not provoking mutant RNA degradation or removal (Dietz et al., Genomics, 1993; Hall and Thein, Blood, 1994). These observations alongside the findings of our study suggests that genetic compensation through mutant defective RNA degradation might be underlying such observations, and further investigation of the phenomenon may help us understand why some mutations cause disease while others do not. It may also lead to the development of more effective therapies for genetic diseases, for example, ones that enhance an individual’s robustness to a mutation rather than correct its effect.
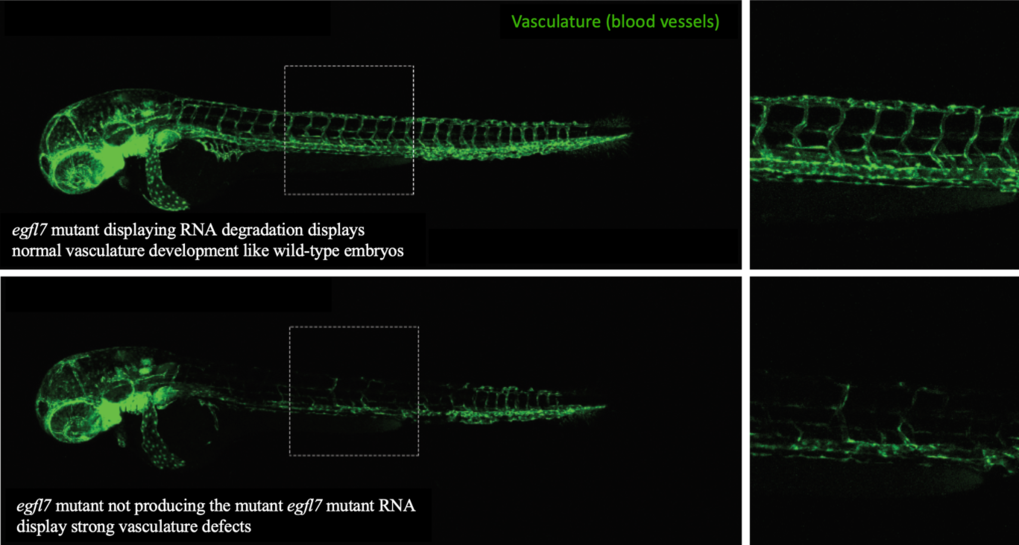
Figure 1: Confocal microscopy images of blood vessels in two different mutant zebrafish larvae for the gene egfl7.
References
Dietz, H. C. et al. Four novel FBN1 mutations: significance for mutant transcript level and EGF-like domain calcium binding in the pathogenesis of Marfan syndrome. Genomics 17, 468–475 (1993).
El-Brolosy, M. A. et al. Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193-197, doi:10.1038/s41586-019-1064-z (2019).
Hall, G. W. & Thein, S. Nonsense codon mutations in the terminal exon of the β-globin gene are not associated with a reduction in β-mRNA accumulation: a mechanism for the phenotype of dominant β-thalassemia. Blood 83, 2031–2037 (1994).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285-291, doi:10.1038/nature19057 (2016).
Sulem, P. et al. Identification of a large set of rare complete human knockouts. Nature genetics 47, 448-452, doi:10.1038/ng.3243 (2015).











